A week ago I attended LessOnline, a rationalist blogging conference featuring many people I’ve known for years—Scott Alexander, Eliezer Yudkowsky, Zvi Mowshowitz, Sarah Constantin, Carl Feynman—as well as people I’ve known only online and was delighted to meet in person, like Joe Carlsmith and Jacob Falkovich and Daniel Reeves. The conference was at Lighthaven, a bewildering maze of passageways, meeting-rooms, sleeping quarters, gardens, and vines off Telegraph Avenue in Berkeley, which has recently emerged as the nerd Shangri-La, or Galt’s Gulch, or Shire, or whatever. I did two events at this year’s LessOnline: a conversation with Nate Soares about the Orthogonality Thesis, and an ask-me-anything session about quantum computing and theoretical computer science (no new ground there for regular consumers of my content).
What I’ll remember most from LessOnline is not the sessions, mine or others’, but the unending conversation among hundreds of people all over the grounds, which took place in parallel with the sessions and before and after them, from morning till night (and through the night, apparently, though I’ve gotten too old for that). It felt like a single conversational archipelago, the largest in which I’ve ever taken part, and the conference’s real point. (Attendees were exhorted, in the opening session, to skip as many sessions as possible in favor of intense small-group conversations—not only because it was better but also because the session rooms were too small.)
Within the conversational blob, just making my way from one building to another could take hours. My mean free path was approximately five feet, before someone would notice my nametag and stop me with a question. Here was my favorite opener:
“You’re Scott Aaronson?! The quantum physicist who’s always getting into arguments on the Internet, and who’s essentially always right, but who sustains an unreasonable amount of psychic damage in the process?”
“Yes,” I replied, not bothering to correct the “physicist” part.
One night, I walked up to Scott Alexander, who sitting on the ground, with his large bald head and a blanket he was using as a robe, resembled a monk. “Are you enjoying yourself?” he asked.
I replied, “you know, after all these years of being coy about it, I think I’m finally ready to become a Rationalist. Is there, like, an initiation ritual or something?”
Scott said, “Oh, you were already initiated a decade ago; you just didn’t realize it at the time.” Then he corrected himself: “two decades ago.”
The first thing I did, after coming out as a Rationalist, was to get into a heated argument with Other Scott A., Joe Carlsmith, and other fellow-Rationalists about the ideas I set out twelve years ago in my Ghost in the Quantum Turing Machine essay. Briefly, my argument was that the irreversibility and ephemerality of biological life, which contrasts with the copyability, rewindability, etc. of programs running on digital computers, and which can ultimately be traced back to microscopic details of the universe’s initial state, subject to the No-Cloning Theorem of quantum mechanics, which then get chaotically amplified during brain activity … might be a clue to a deeper layer of the world, one that we understand about as well as the ancient Greeks understood Newtonian physics, but which is the layer where mysteries like free will and consciousness will ultimately need to be addressed.
I got into this argument partly because it came up, but partly also because this seemed like the biggest conflict between my beliefs and the consensus of my fellow Rationalists. Maybe part of me wanted to demonstrate that my intellectual independence remained intact—sort of like a newspaper that gets bought out by a tycoon, and then immediately runs an investigation into the tycoon’s corruption, as well as his diaper fetish, just to prove it can.
The funny thing, though, is that all my beliefs are the same as they were before. I’m still a computer scientist, an academic, a straight-ticket Democratic voter, a liberal Zionist, a Jew, etc. (all identities, incidentally, well-enough represented at LessOnline that I don’t even think I was the unique attendee in the intersection of them all).
Given how much I resonate with what the Rationalists are trying to do, why did it take me so long to identify as one?
Firstly, while 15 years ago I shared the Rationalists’ interests, sensibility, and outlook, and their stances on most issues, I also found them bizarrely, inexplicably obsessed with the question of whether AI would soon become superhumanly powerful and change the basic conditions of life on earth, and with how to make the AI transition go well. Why that, as opposed to all the other sci-fi scenarios one could worry about, not to mention all the nearer-term risks to humanity?
Suffice it to say that empirical developments have since caused me to withdraw my objection. Sometimes weird people are weird merely because they see the future sooner than others. Indeed, it seems to me that the biggest thing the Rationalists got wrong about AI was to underestimate how soon the revolution would happen, and to overestimate how many new ideas would be needed for it (mostly, as we now know, it just took lots more compute and training data). Now that I, too, spend some of my time working on AI alignment, I was able to use LessOnline in part for research meetings with colleagues.
A second reason I didn’t identify with the Rationalists was cultural: they were, and are, centrally a bunch of twentysomethings who “work” at an ever-changing list of Berkeley- and San-Francisco-based “orgs” of their own invention, and who live in group houses where they explore their exotic sexualities, gender identities, and fetishes, sometimes with the aid of psychedelics. I, by contrast, am a straight, monogamous, middle-aged tenured professor, married to another such professor and raising two kids who go to normal schools. Hanging out with the Rationalists always makes me feel older and younger at the same time.
So what changed? For one thing, with the march of time, a significant fraction of Rationalists now have marriages, children, or both—indeed, a highlight of LessOnline was the many adorable toddlers running around the Lighthaven campus. Rationalists are successfully reproducing! Some because of explicit pronatalist ideology, or because they were persuaded by Bryan Caplan’s arguments in Selfish Reasons to Have More Kids. But others simply because of the same impulses that led their ancestors to do the same for eons. And perhaps because, like the Mormons or Amish or Orthodox Jews, but unlike typical secular urbanites, the Rationalists believe in something. For all their fears around AI, they don’t act doomy, but buzz with ideas about how to build a better world for the next generation.
At a LessOnline parenting session, hosted by Julia Wise, I was surrounded by parents who worry about the same things I do: how do we raise our kids to be independent and agentic yet socialized and reasonably well-behaved, technologically savvy yet not droolingly addicted to iPad games? What schooling options will let them accelerate in math, save them from the crushing monotony that we experienced? How much of our own lives should we sacrifice on the altar of our kids’ “enrichment,” versus trusting Judith Rich Harris that such efforts quickly hit a point of diminishing returns?
A third reason I didn’t identify with the Rationalists was, frankly, that they gave off some (not all) of the vibes of a cult, with Eliezer as guru. Eliezer writes in parables and koans. He teaches that the fate of life on earth hangs in the balance, that the select few who understand the stakes have the terrible burden of steering the future. Taking what Rationalists call the “outside view,” how good is the track record for this sort of thing?
OK, but what did I actually see at Lighthaven? I saw something that seemed to resemble a cult only insofar as the Beatniks, the Bloomsbury Group, the early Royal Society, or any other community that believed in something did. When Eliezer himself—the bearded, cap-wearing Moses who led the nerds from bondage to their Promised Land in Berkeley—showed up, he was argued with like anyone else. Eliezer has in any case largely passed his staff to a new generation: Nate Soares and Zvi Mowshowitz have found new and, in various ways, better ways of talking about AI risk; Scott Alexander has for the last decade written the blog that’s the community’s intellectual center; figures from Kelsey Piper to Jacob Falkovich to Aella have taken Rationalism in new directions, from mainstream political engagement to the … err … statistical analysis of orgies.
I’ll say this, though, on the naysayers’ side: it’s really hard to make dancing to AI-generated pop songs about Bayes’ theorem and Tarski’s definition of truth not feel cringe, as I can now attest from experience.
The cult thing brings me to the deepest reason I hesitated for so long to identify as a Rationalist: namely, I was scared that if I did, people whose approval I craved (including my academic colleagues, but also just randos on the Internet) would sneer at me. For years, I searched of some way of explaining this community’s appeal so reasonable that it would silence the sneers.
It took years of psychological struggle, and (frankly) solidifying my own place in the world, to follow the true path, which of course is not to give a shit what some haters think of my life choices. Consider: five years ago, it felt obvious to me that the entire Rationalist community might be about to implode, under existential threat from Cade Metz’s New York Times article, as well as RationalWiki and SneerClub and all the others laughing at the Rationalists and accusing them of every evil. Yet last week at LessOnline, I saw a community that’s never been thriving more, with a beautiful real-world campus, excellent writers on every topic who felt like this was the place to be, and even a crop of kids. How many of the sneerers are living such fulfilled lives? To judge from their own angry, depressed self-disclosures, probably not many.
But are the sneerers right that, even if the Rationalists are enjoying their own lives, they’re making other people’s lives miserable? Are they closet far-right monarchists, like Curtis Yarvin? I liked how The New Yorker put it in its recent, long and (to my mind) devastating profile of Yarvin:
The most generous engagement with Yarvin’s ideas has come from bloggers associated with the rationalist movement, which prides itself on weighing evidence for even seemingly far-fetched claims. Their formidable patience, however, has also worn thin. “He never addressed me as an equal, only as a brainwashed person,” Scott Aaronson, an eminent computer scientist, said of their conversations. “He seemed to think that if he just gave me one more reading assignment about happy slaves singing or one more monologue about F.D.R., I’d finally see the light.”
The closest to right-wing politics that I witnessed at LessOnline was a session, with Kelsey Piper and current and former congressional staffers, about the prospects for moderate Democrats to articulate a pro-abundance agenda that would resonate with the public and finally defeat MAGA.
But surely the Rationalists are incels, bitter that they can’t get laid? Again, the closest I saw was a session where Jacob Falkovich helped a standing-room-only crowd of mostly male nerds confront their fears around dating and understand women better, with Rationalist women eagerly volunteering to answer questions about their perspective. Gross, right? (Also, for those already in relationships, Eliezer’s primary consort and former couples therapist Gretta Duleba did a session on relationship conflict.)
So, yes, when it comes to the Rationalists, I’m going to believe my own lying eyes over the charges of the sneerers. The sneerers can even say about me, in their favorite formulation, that I’ve “gone mask off,” confirmed the horrible things they’ve always suspected. Yes, the mask is off—and beneath the mask is the same person I always was, who has an inordinate fondness for the Busy Beaver function and the complexity class BQP/qpoly, and who uses too many filler words and moves his hands too much, and who strongly supports the Enlightenment, and who once feared that his best shot at happiness in life would be to earn women’s pity rather than their contempt. Incorrectly, as I’m glad to report. From my nebbishy nadir to the present, a central thing that’s changed is that, from my family to my academic colleagues to the Rationalist community to my blog readers, I finally found some people who want what I have to sell.
Unrelated Announcements:
My replies to comments on this post might be light, as I’ll be accompanying my daughter on a school trip to the Galapagos Islands!
A few weeks ago, I was “ambushed” into leading a session on philosophy and theoretical computer science at UT Austin. (I.e., asked to show up for the session, but thought I’d just be a participant rather than the main event.) The session was then recorded and placed on YouTube—and surprisingly, given the circumstances, some people seemed to like it!
Friend-of-the-blog Alon Rosen has asked me to announce a call for nominations for a new theoretical computer science prize, in memory of my former professor (and fellow TCS blogger) Luca Trevisan, who was lost to the world too soon.
And one more: Mahdi Cheraghchi has asked me to announce the STOC’2025 online poster session, registration deadline June 12; see here for more. Incidentally, I’ll be at STOC in Prague to give a plenary on quantum algorithms; I look forward to meeting any readers who are there!







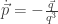

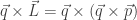


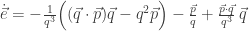
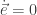
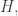


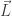

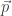

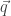


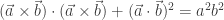
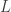



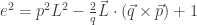




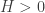

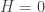


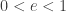







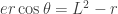
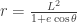
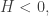
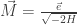
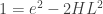
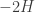

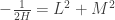



.")





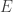 will show up with probability proportional to
will show up with probability proportional to  in equilibrium at temperature
in equilibrium at temperature 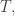 where
where 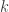 is Boltzmann’s constant. But dynamics is much harder, so the Arrhenius equation for the rates of transitions between states is precious, even though only approximate. I would like to understand this law better, and its range of approximate validity. The above paper digs into that. As an example, it studies the rate at which Stone–Wales transformations happen in buckminsterfullerene!
is Boltzmann’s constant. But dynamics is much harder, so the Arrhenius equation for the rates of transitions between states is precious, even though only approximate. I would like to understand this law better, and its range of approximate validity. The above paper digs into that. As an example, it studies the rate at which Stone–Wales transformations happen in buckminsterfullerene!



































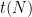 studied in this paper, allowing us to settle all the previous conjectures about this quantity in the literature.
studied in this paper, allowing us to settle all the previous conjectures about this quantity in the literature.
 such that the factorial
such that the factorial  can be expressed as a product of
can be expressed as a product of 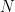 factors, each of which is at least
factors, each of which is at least  ; we have been able to compute
; we have been able to compute  . In fact, we can get surprisingly sharp upper and lower bounds on
. In fact, we can get surprisingly sharp upper and lower bounds on 
 , which we conjecture to be improvable to
, which we conjecture to be improvable to
 : … For instance, we can demonstrate numerically that
: … For instance, we can demonstrate numerically that


 for all
for all  .
.  for all
for all  .
.  for all
for all  . (In fact we show this is true for
. (In fact we show this is true for  , and that this threshold is best possible.)
, and that this threshold is best possible.)  for all large
for all large  and
and 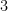 from the standard factorization
from the standard factorization  of
of  :
: 
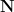 . In the Lean formalization, I first develop “by hand” an alternate construction
. In the Lean formalization, I first develop “by hand” an alternate construction 
 , where
, where 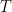 is a parameter going to infinity,
is a parameter going to infinity,  counts the number of zeroes of the Riemann zeta function of real part at least
counts the number of zeroes of the Riemann zeta function of real part at least  and imaginary part between
and imaginary part between 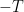 and
and  is an exponent which one would like to be as small as possible. The Riemann hypothesis would allow one to take
is an exponent which one would like to be as small as possible. The Riemann hypothesis would allow one to take  for any
for any  , but this is an unrealistic goal, and in practice one would be happy with some non-trivial upper bounds on
, but this is an unrealistic goal, and in practice one would be happy with some non-trivial upper bounds on  for all
for all  ); this hypothesis is currently known for
); this hypothesis is currently known for  and
and  , but the known bounds are not strong enough to establish this hypothesis in the remaining region. However, there was a recent advance of Guth and Maynard, which among other things improved the upper bound
, but the known bounds are not strong enough to establish this hypothesis in the remaining region. However, there was a recent advance of Guth and Maynard, which among other things improved the upper bound 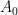 on
on  from
from  to
to  , marking the first improvement in this bound in over four decades. Here is a plot of the best known upper bounds on
, marking the first improvement in this bound in over four decades. Here is a plot of the best known upper bounds on 
 for all
for all  holding for all
holding for all  if
if  , and for almost all
, and for almost all  . For instance, the Guth-Maynard results give a prime number theorem in almost all short intervals for
. For instance, the Guth-Maynard results give a prime number theorem in almost all short intervals for 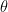 as small as
as small as  , and the density hypotheis would lower this just to
, and the density hypotheis would lower this just to  .
.
 , which roughly speaking amounts to getting an upper bound of
, which roughly speaking amounts to getting an upper bound of  on the size of the exceptional set in any large interval
on the size of the exceptional set in any large interval ![{[X,2X]}](https://s0.wp.com/latex.php?latex=%7B%5BX%2C2X%5D%7D&bg=ffffff&fg=000000&s=0&c=20201002) . Based on the above assertions, one expects
. Based on the above assertions, one expects 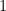 for
for  , be bounded by
, be bounded by  for
for  , but have some intermediate bound for the remaining exponents.
, but have some intermediate bound for the remaining exponents.
 of prime gaps) by such authors as
of prime gaps) by such authors as  where
where  Actually, by also utilizing fourth moment methods, we obtain a stronger bound
Actually, by also utilizing fourth moment methods, we obtain a stronger bound  where
where  and
and  is the exponent in “additive energy zero density theorems”
is the exponent in “additive energy zero density theorems”  where
where  is similar to
is similar to 



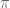 , which is also known as Archimedes’ constant: the
, which is also known as Archimedes’ constant: the 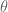 really called the angle of declination?
really called the angle of declination? , one can approximately write the relationship
, one can approximately write the relationship  between the mountain height
between the mountain height 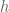 , the Earth radius
, the Earth radius 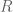 , and the dip angle
, and the dip angle  . The key point here is the inverse quadratic dependence on
. The key point here is the inverse quadratic dependence on  radians, leading to an estimate of
radians, leading to an estimate of 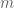 and
and 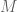 . If one takes the logarithm of the inverse square law in the video, and performs the normalizations used by astronomers to define magnitude, one arrives at the standard relation
. If one takes the logarithm of the inverse square law in the video, and performs the normalizations used by astronomers to define magnitude, one arrives at the standard relation  between absolute and apparent magnitude.
between absolute and apparent magnitude.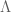 , known as the
, known as the  of the flatness of the universe which, for reasons still not fully understood (but likely caused by some sort of inflation mechanism), happens to be extremely close to zero. As such, predictions for the size of the universe remain highly volatile at the current level of measurement accuracy.
of the flatness of the universe which, for reasons still not fully understood (but likely caused by some sort of inflation mechanism), happens to be extremely close to zero. As such, predictions for the size of the universe remain highly volatile at the current level of measurement accuracy. of a particle, which may take two values,
of a particle, which may take two values, 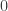 and
and 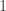 . Assumption 1 tells us that if Alice performs this measurement when the particle is in a superposition state, the joint system of Alice’s brain and the particle will end up in an entangled state. Now Alice’s mind-substrate is not in a pure state, so by Assumption 3 does not have a definite experience. This contradicts Assumption 2. Wigner’s proposed resolution to this paradox is that in fact Assumption 1 is incorrect, and that there is an influence of the mental on the physical, namely objective collapse or, as he puts it, that the “statistical element which, according to the orthodox theory, enters only if I make an observation enters equally if my friend does”.
. Assumption 1 tells us that if Alice performs this measurement when the particle is in a superposition state, the joint system of Alice’s brain and the particle will end up in an entangled state. Now Alice’s mind-substrate is not in a pure state, so by Assumption 3 does not have a definite experience. This contradicts Assumption 2. Wigner’s proposed resolution to this paradox is that in fact Assumption 1 is incorrect, and that there is an influence of the mental on the physical, namely objective collapse or, as he puts it, that the “statistical element which, according to the orthodox theory, enters only if I make an observation enters equally if my friend does”. at time
at time 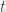 .” Then agent A can conclude that “I am certain that
.” Then agent A can conclude that “I am certain that  at time
at time 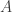 and
and 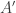 are certain about their respective judgments, since these states of knowledge change.
are certain about their respective judgments, since these states of knowledge change.





{kind=link}
{kind=link}